In the last section, we explored the ARAGORN command line program and how the
file-oriented paradigm it follows complicates its usage. Now we will look into
the ARAGORN source code and see what it took to refactor it into a pure
library. In the next section, we will see how to lift this pure library into a
morloc module and then how to incorporate it into powerful new programs.
The Aragorn program is written as a single 12000-line C file with dependencies
only on stdio and stdlib. The code is undocumented, but the style is
reasonably clear with little gratuitous obscurity or excess frameworking. The
code makes heavy use of pointer arithmetic, single-letter variable names, and
GOTO statements. They never use logical AND, rather opting for nested
if-statements. The programmer learned enough C to express their intent and
ignored modern norms – I can respect that.
Most of the code is wavy lines of conditions speckled with many mutating pointers into a sequence. Here is one short function as an example:
void overlap(data_set *d, int sort[], int n, int it, csw *sw)
{ int i,j,flag,cross,crosstoo;
long a,b,e,f,a2,b2,e2,f2,psmax;
char sname[100],s[100];
flag = 0;
cross = 0;
psmax = d->psmax;
a = ts[it].start;
b = ts[it].stop;
if (b < a)
{ a2 = a - psmax;
b2 = b;
b += psmax;
cross = 1; }
j = -1;
while (++j < n)
{ i = sort[j];
if (i == it) continue;
e = ts[i].start;
f = ts[i].stop;
crosstoo = 0;
if (f < e)
{ e2 = e - psmax;
f2 = f;
f += psmax;
crosstoo = 1; }
if (a <= f)
if (b >= e)
goto OV;
if (crosstoo)
if (a <= f2)
if (b >= e2)
goto OV;
if (cross)
{ if (a2 <= f)
if (b2 >= e)
goto OV;
if (crosstoo)
if (a2 <= f2)
if (b2 >= e2)
goto OV; }
continue;
OV:
if (!flag) fputc('\n',sw->f);
name(ts+i,sname,1,sw);
location(s,ts+i,sw,sname);
fprintf(sw->f,"Overlap with %d: %s\n", j+1,s);
flag = 1; }
if (flag) fputc('\n',sw->f); }There are few relevant particulars. First, notice the print statements at the
end of the function. IO and algorithmic logic are interwoven throughout the
program. Second, the return type is void. Nothing is returned. Instead the
function is run for its side effects. One of these effects is the print
statements. Three mutable values are passed into the function (d, sort,and
sw). Skimming the code I don’t see any place where these arguments actually
are mutated. But there is also a global value, ts, which is an array of
observed tRNAs, that is mutated in the location function.
Without documentation, I am left to guess what much of the code does. For example, the matrix below:
int gc[6][6] = { { 0,0,0,0,0,0 },
{ 0,0,1,0,1,0 },
{ 0,1,0,0,1,0 },
{ 0,0,0,0,0,0 },
{ 0,1,1,0,1,0 },
{ 0,0,0,0,0,0 } };I know that input sequence is mapped to 6 values: the four nucleotides (A, C, G,
T), an unknown base (N), and an invalid character. A 6-by-6 matrix would map
pairs of bases to one another. This matrix is symmetric, is named gc, and maps
pairs of bases to binary values. G maps to C and N maps to G or C. Altogether,
this matrix appears to be designed to detect GC pairs. From this code we learn
that any pairing that includes an unknown character will match the GC
pattern. This is a very lenient choice.
Similarly, the matrix below recognizes canonical and wobble (G-T) base pairs (along with any pairing that includes an N):
int bp[6][6] = { { 0,0,0,1,1,0 },
{ 0,0,1,0,1,0 },
{ 0,1,0,1,1,0 },
{ 1,0,1,0,1,0 },
{ 1,1,1,1,1,0 },
{ 0,0,0,0,0,0 } };The matrices are used in code like the following:
s1 = sc;
s2 = sc + 16;
sd = sc + 6;
j = bp[*s1][*--s2];
while (++s1 < sd) j += bp[*s1][*--s2];
if (j >= 6) { /* handle matched D-stem */ }This code recognizes a hairpin. sc is pointer to a current search location in
the sequence. j is a count of the number of base-pair matches. s1 and s2
are pointers to the potential start and stop of the hairpin loop. The ++s1 and
--s2 operations are pre-increment and pre-decrement operations. These mutate
the pointers, shifting to the next pair of bases in the stem, and return the new
values to the expression. The while loop counts the number of matches in the
stem. If six stem bases match, then the if condition is met and the block for
a matched D-stem is executed.
stem_1 loop stem_2
------ ------
xxx.................xxxxx
^ ^ ^
sc sd sc + 16
^ ^
s1 --> <-- s2
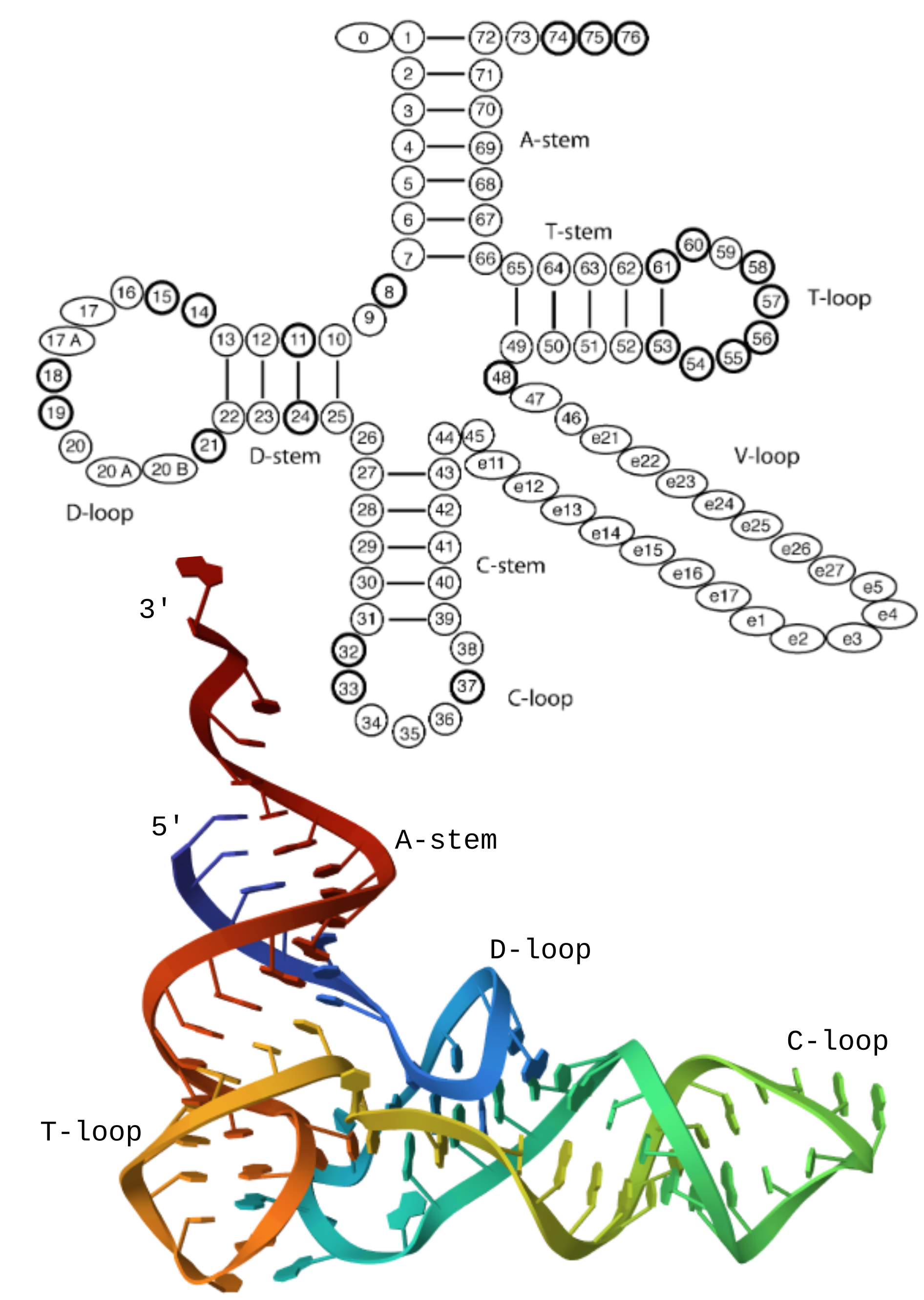 tRNA molecular structure. The top diagram shows the components of a tRNA and
the canonical numbering (from the Laslett2004 paper, Figure 1). The colored
image below shows the crystal structure of yeast phenylalanine tRNA
(1EHZ). The 5’ end is blue and transforms to red at the 3’. Image made on PDB website.
tRNA molecular structure. The top diagram shows the components of a tRNA and
the canonical numbering (from the Laslett2004 paper, Figure 1). The colored
image below shows the crystal structure of yeast phenylalanine tRNA
(1EHZ). The 5’ end is blue and transforms to red at the 3’. Image made on PDB website.
ARAGORN detects tRNAs by first searching for the strong GTTC pattern in the T-loop (indices 53-56 in the figure on the right). Then the pair matching bases in the T-loop stem are sought. If they are found a reasonable distance from the end of the GTTC pattern, then a T-loop hit is returned. Then, anchoring at the T-loop position, the remainder of the tRNA pattern is searched for. The relative rarity of the T-loop pattern allows must locations in the search sequence to be skipped, avoiding the expensive search for the A-stem and possible introns in the C-loop.
When a complete tRNA is found, the energy is checked against the minimum permitted energy. If the score is high enough, the tRNA is stored. After the input has been processed, tRNAs that overlap other tRNAs and have a lower score may be removed (depending on input flags). Then the remaining tRNAs are printed to standard output in the format specified by the user.
Refactor
The goal of this refactor is to create a program that compiles to a shared library that exports a single pure function transforming a single DNA string to a list of predicted tRNAs.
There are several concerns that the new library will NOT handle, these are listed below:
Output formatting. The function will return only a list of tRNAs. These tRNA descriptions will contain the information needed for downstream functions to create any relevant representation. It is not the role of the base library to anticipate all use cases. In the next section, I will create a second module for many opinionated options for exploring the tRNAs. Others may develop alternative modules. My goal is to decouple the fundamental scientific algorithm (tRNA prediction) from downstream applications.
Error handling and input validation. I will preserve the original ARAGORN leniency to invalid input sequence. Presence of invalid characters in a region where we are predicting a tRNA will trigger a negative match at that site and the algorithm will proceed to the next site. There is nothing about sequence validation that is unique to ARAGORN. All sequence-based tools require input validation. A given sequence may be used in one pipeline by many functions, if each function validates it, then we are repeating work. This repetition wastes CPU time and requires code repetition across tools. It is better to validate the sequence in a dedicated general-purpose function and allow all sequence-based tools to assume the input sequence is valid. So ARAGORN will have no need to handle errors.
Logging. As a pure function, the tRNA predictor will not have any side effects. This includes printing logging messages. If logging is required, it can be provided by an outside functions. I will explore solutions to parallelizing tRNA search across a single large sequence (e.g., a eukaryotic chromosome which may be larger than 100Mb). If desired, progress logging could be added to whatever function handles the parallelization process. Alternatively, in the future,
morlocitself will be able to automatically generate many forms of progress logging.Species-specific handling. The function sees only a sequence. It does not know where the sequence came from. I am open to changing this if there is good reason. It may be reasonable to support different functions for mitochondrial, eukaryotic, and bacterial tRNAs. But for now, I will keep the function general. This means that translation of the codon cannot be performed since the gene code is not known. Translation is a complex process that requires a curated list of gene codes and a means to map species to gene code. It is best delegated to a dedicated module. I will cover this in the next section.
In the refactor, I strip away all the code related to the above concerns.
The refactor reduced the code base from 12439 lines to just 1006. Lines of code is a poor metric for complexity. Some of these reduced lines do not represent meaningful drops in complexity. Major such deletions include the 677-line license (I moved this to a dedicated file), the 580-line list of amino acid tables and the 1276-line list of species-specific tag peptides (used in the tmRNA code). I also added documentation and less compact spacing, increasing the refactor line count.
Better complexity metrics include Halstead Volume (a measure of vocabulary) and cyclomatic complexity (a measure of branching). Since I don’t have a tool handy for measuring these metrics, I’ll spin my own metric – the if-count. This is a direct count of the number of times the code branches on a condition. The following little one-liner does the job:
$ grep -c "if *(" aragorn1.2.41.cThe original code had 2547 if statements and the refactored code dropped to just 99. I can randomly spot check the results:
$ grep "if *(" aragorn/src/aragorn1.2.41.c | sort -R | head
else if (c3 == 'F') { sw.ifixedpos = 1; s++; lv--; }
if (tstem >= 5)
do if ((ic = (int)fgetcd(d)) == NOCHAR) goto FAIL;
if ((dhit[nd].bondtype & 0xf) < 1)
if (RI[cloopend[-2]])
if (var <= 7)
if (bp[cend[k]][b57]) energy += 1.0; }}
while (se < tpos) if (*se++ == Thymine) k++;
if (((mabondtype & 0xf) > 3) || (bondtype < 0x1000))
if (w & 1) s = copy("amino acceptor",s);The pattern may be simplistic, but it looks like I am roughly grepping out the right lines.
Next I will break down where the most code was removed. I partitioned functions in the source code into 6 categories: definitions, UI, input, output, tmRNA, and core. The lines of code, if-count and number of functions are listed in the table below:
| Category | LOC | if-count | functions |
|---|---|---|---|
| UI: argument parsing and main | 576 | 107 | 6 |
| definitions: structs and constants | 2662 | 0 | 0 |
| input: FASTA and GenBank parsing | 1121 | 298 | 23 |
| output: raw, fasta, batch, SVG | 2495 | 421 | 58 |
| tmRNA: pattern matching | 3688 | 1498 | 11 |
| tRNA: pattern matching | 1087 | 224 | 22 |
| refactored: complete tRNA code | 1006 | 99 | 10 |
The comparison is weakened since tmRNA is not yet supported.
The program also originally had no dynamic data structures. For example, to store inferred tRNAs, a single array of 200 elements is allocated. As new tRNAs are predicted, they are set to elements in the array. If the array index reaches 200, then an error is raised.
This was a massive project that required almost a month of my limited spare time. The code was undocumented and heavily reliant on pointer arithmetic, mutation, and cryptic variable naming.
The risk of introducing bugs during this refactor was high. So I assembled a large set of test data. I need to ensure that the final algorithm is both correct and as fast as the original.
The choice to use pure C has its perks, including portability between systems and very fast compile times. But I’ve moved to C++ by replacing static data structures with vectors and strings from the standard template library.
I have always argued that morloc frees the programmer to write simpler
code. Within the morloc ecosystem, a scientific programmer need only write the
core algorithms of scientific interest. They can ignore parsing and formatting,
user interfaces, parallelism (usually), metadata propagation, and similar
complexities. In this section, I will show how this works in practice.
A new data representation
What data should be returned? First, the RNA type and anticodon are certainly
needed (e.g., tRNA-Arg and tct). Second, the location is needed (the
sequence ID and start and stop indices). Third, the inferred structure may
optionally be returned as a connectivity matrix. There may be additional values
that would be useful to return, such as some measure of statistical
significance, the melting points of various structures, the G/C content, and
such.
The input is a configuration record and a sequence. So the basic type signatures will be something like this:
data AragornConfig = AragornConfig {
...
}
findTRNA :: AragornConfig -> DNA -> [(RNAType, Start, Stop, Anticodon, AminoAcid, Matrix)] void bopt_fastafile(data_set *d, csw *sw)
{ int i,nt,flag,len;
int *s,*sf,*se,*sc,*swrap;
int seq[2*LSEQ+WRAP+1],cseq[2*LSEQ+WRAP+1],wseq[2*WRAP+1];
long gap,start,rewind,drewind,psmax,tmaxlen,vstart,vstop;
double sens;
FILE *f = sw->f;
rewind = MAXTAGDIST + 20;
if (sw->trna | sw->mtrna)
{ tmaxlen = MAXTRNALEN + sw->maxintronlen;
if (rewind < tmaxlen) rewind = tmaxlen; }
if (sw->tmrna)
if (rewind < MAXTMRNALEN) rewind = MAXTMRNALEN;
if (sw->peptide)
if (sw->tagthresh >= 5)
if (rewind < TSWEEP) rewind = TSWEEP;
sw->loffset = rewind;
sw->roffset = rewind;
drewind = 2*rewind;
d->ns = 0;
d->nf = 0;
d->nextseq = 0L;
d->nextseqoff = 0L;12241 12240 test-trna_sequence_cmp_arc_1.txt 958811 958748 test-trna_sequence_cmp_bac_1.txt 2188 2188 test-trna_sequence_fungi_1.txt 13919 13897 test-trna_sequence_phage_1.txt 1352 1350 test-trna_sequence_plant_1.txt 3852 3850 test-trna_sequence_plasmid_1.txt 974 974 test-trna_sequence_virus_1.txt
This not-so-simple picture is further complicated by the fact that tRNAs may have introns. Strangely, these introns are not spliced out by the same mechanisms that splice mRNAs. Introns may be present in tRNA bacteria, not just Eukaryotes.
tRNA introns (Schmidt 2019)
CCA acceptor stem - this may be encoded in the tRNA itself or it may be added in a post-processing step.
`Aragorn is reasonably tolerant of malformed input. All bytes are translated into either DNA bases, unknowns, or ambiguous bases.